
You have no items in your shopping cart
An Introduction to Distillation
- Posted on
- By Goldleaf Scientific
- Posted in Buyers Guides, How To Guides, Short Path Distillation
- 2
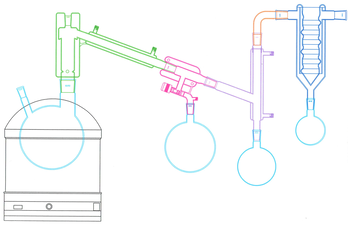
Have you ever wondered by there’s morning dew or why your breath fogs a window? We often think as a vapor as steam, coming off from a boiling pot, but just like some hot soup the steam does not have to come from a boiling liquid. Liquids will exert a vapor pressure at temperatures well below their boiling point. A boiling point is merely the temperature at which the vapor pressure of the liquid is greater than the pressure of the atmosphere. This is why water boils at different temperatures when at different altitudes.
When the day is warm there is lots of water vapor in the air, but as the night cools the air the water vapor condenses on surfaces. This is the reason behind the morning dew and why cold air is so dry. The lower the temperature the lower the vapor pressure.
We must first begin by understanding Raoult’s law which states that the vapor pressure of a compound is the sum of the vapor pressures of all compounds times by its mole fraction (or the ratio of it over the sum of all other compounds). In this case we look at two compounds A, and B, but a real life mixture often has many more.

Suppose that compound A is ethanol and compound B is water. Ethanol has a higher vapor pressure than water so if we had an equal ratio of water and ethanol the ethanol would have a higher vapor pressure and would be enriched in the vapor phase. By boiling the equal mixture the vapor will have a greater ratio of ethanol to water, but water is also present in the vapor phase as well so we must take that into consideration. A pure sample of ethanol will boil at 178°F while pure water will boil at 212°F, and a mixture will boil at some point in between those. By finding the value for vapor pressure (an intrinsic value that can be found online) and the mole fraction of each component you can estimate the boiling point and the ratio of compounds in the vapor phase.
Fractional Distillation:
When a batch of volatile liquid needs to be separated into its components, we might simply assume that the distillation process would provide the individual components in pure form. No simple distillation can produce this magic. A binary mixture, under the best of conditions, will produce component A of some purity, component B of similar purity, and a mid-fraction that closely resembles the starting mixture. The degree of purity desired in the 'pure' products will determine the size of the mid-fraction.

Simple distillation, consisting of a heated boiling pot, a thermocouple (or thermometer), a condenser and a receiver will be barely capable of separating materials that have differences in boiling point of less than ~60°C. Here's a rough idea of how this goes:

Small boiling point differences lead to practically no separation, while only large differences provide some separation. Simple distillation is only useful for removing low boiling materials from solids or very high boiling materials. To obtain good separations, a fractionation column is required. Simply put a fractional column serves to allow the vapor to condense and be redistilled by the incoming vapor as it rises through the column. In our short path distillation setup the distillation head has a short section with vigreux teeth that serve this purpose.
Reflux Ratio:
The fractional column can only improve the separation of components if vapor is continually being condensed and draining back through the column to the distillation flask. This allows higher boiling material to be 'washed' back down to the distillation flask, simultaneously causing an enrichment of lower boiling material in the takeoff vapor. The ratio of condensed vapor returning to the column to the condensed vapor taken off as product is called the reflux ratio.
The reflux ratio will drastically affect the setup's separation capability. To get a reasonable split of two components that differ in BP by only 5°C, you would need a 24"well packed column, and about 20 drops of distillate going back to the column for every drop you are able to harvest! Expect a 600 ml starting batch to take several days to separate. At the end, you will have 200ml of 95% purity A, 200ml of mid fraction that's essentially the same composition as the starting material and 150ml of 95% purity B. The remaining 50 ml will be sizzling at the bottom of the pot and 'held-up' in the column, stuck to the packing.
Column Hold-up:
This is the difference in temperature between the two boiling points
With no packing, the Vigreux column has perhaps the lowest holdup, but it's separation efficiency is not as great as a very long packed column which will obviously hold up more than a short one. A long column, worked at a low reflux ratio, will deliver quicker results, but hold up more product than a short column worked at a high reflux ratio.

Column Flooding:
To conduct a fractional distillation most efficiently in terms of operation time, the takeoff rate (how much is being condensed for removal) must be as high as possible, yet to achieve a good separation, the reflux ratio must be adequate. If the rate of rising vapor, against the rate of falling liquid becomes too great, the column will flood. Flooding is easily observed as excess liquid within the column packing that is bubbling or boiling. In a normally operated column there will be evidence of liquid drops occasionally falling through the packing, but it will be quite peaceful in general. The moment you see liquid getting blown upwards, even in the smallest way, the column is beginning to flood.
This is one good advantage to using vacuum jacketed columns, as they do not dissipate as much heat and allow you to easily view the column. Hydrocarbons in general and small alcohols have very low viscosity, and can be 'pushed' to very high power levels, but viscous liquids can flood a column very quickly and unexpectedly. Viscous materials do not drain through the column quickly; they 'ooze' back down the column, and close the spaces required for the rising vapor flow. A flooded column is a useless column and it is crucial to always start fractionations at a low temperature level and slowly advance the temperature with a keen eye toward flooding and reflux ratio. If flooding is noticed, back the power by some 20% or so and leave it there for a while.
Spotting a New Fraction:
We know that water boils at 100°C or 212°F, but what if we took that water and poured it onto a surface that was as hot as the sun the water would boil, but at what temperature?
100°C. That’s right. No matter how hot we expose the water to it will always boil at 100°C. More of it will boil quicker is all. It might be so hot that it instantaneously turns it to water vapor but it still boiled at 100°C. I make this point because as you distill your substance it will keep a steady temperature until it’s done and then will rise in temperature. It’s not quite as easy as spotting the perfectly horizontal spots in a temperature vs time chart as the picture below indicates, there are some things to keep in mind, but in general that’s how it works. Each fraction boils at a steady temperature until there is no more, then rises in temperature to the boiling point of the new fraction and so on.

The chart on the right is a more realistic temperature vs time curve. The straight red lines indicate a constant fraction is being distilled. Notice that there is still somewhat of a slope (although it is exaggerated in the picture). This is because of Raoult’s law and the fact that all compounds in the mixture exert a partial pressure. You should look for is when the temperature vs time slope begins to change. As the curve starts to diverge from the straight red line this indicate a new fraction is starting. Mathematically stated when the curve becomes nonlinear it indicates a new fraction has begun to be distilled.
All references to temperature here will be assumed to be taken from the thermometer in the distillation head. Only take temperature readings from this source.
It is a useful tool to create a chart so that you can find the ideal moment to collect a new fraction. This can be done with a software program paired with the digital temperature controller on the hotplate stirrer, but you can also do this with just a pencil and some graphing paper. As previously stated, start or stop to collect a fraction before getting into the danger zone. This makes things much less stressful because if you mess up you won’t have to redistill the entire sample. Start in the bottom left hand side of the graphing paper.
Each line up represents a degree higher temperature, and each line left represents another minute of time. Every minute take a new temperature reading a mark it on the graphing paper. After several readings simply connect the dots. Use a ruler to draw a straight line with the trend of the data points and when you start to see the chart separate from the line you drew with the ruler then it’s time to collect a new fraction. An important point is that your heating mantle will have a temperature swing. This is the temperature range in which the hotplate stirrer turns on and off. The greater the swing, the greater the temperature fluctuation in your readings. It is therefore wise to set your swing value at the minimal level. This is important because your readings will also swing and it’s important not to confuse these as an indication of a new fraction.
You can always simply cut your fraction early/late before it gets ruined by the next fraction. This will ensure that the work you have done up to that point is safe and if you ruin the next fraction it will be a much smaller amount that you can always redistill at a later point. For example if you were short path distilling a botanical extract and don’t want the terpenes (which foul) into your extract then you would want to collect all of the terpenes and sacrifice some of your desired fraction yield by letting it carry over until you are confident that no more terpenes are there. Of course if your do not fully get full separation by rushing or being impatient then it will be of no use. You must apply correct technique.
Tip: You can visually see the difference in viscosity between terpenes and your more viscous plant compounds as they condense.
A FINAL THOUGHT
Notes: Always have the stillpot stirred by a magnetic stirbar. The tendency for materials to 'bump' can be extreme, and can cause actual damage (breakage), as well as poor fractionation. A stirbar also leaves local pressure variations in its wake as it stirs, giving rise to bubbles before the liquid overheats.
NEVER use a cracked (even tiny ones) glassware ESPECIALLY when under vacuum. This is a serious safety issue. Visually inspect each glass component under bright light before each use. Inspect at varying angle to the light source and replace any suspect glassware before using. It may be convenient to keep spare parts for such an occasion.
SOMETHING TO CONSIDER:
While the fractionation is approaching a split point, and you've got nothing to do but watch the slowly dripping distillate, consider the state of the pot containing nearly 100% B component, with just a few A molecules left. How do the A molecules escape to the receiver and leave the B molecules behind? Answer is, they can't, without carrying B molecules with them. That's why you'll never get a totally pure distillate from a mixture. This is also why so much reflux is required; consider how many times the entire volume of the material in the stillpot needs to be completely vaporized and cycled through the column to get those few remaining molecules of A out! Since the entire volume cannot be vaporized at once, and pushed through the magic fractionating process, each full volume of stillpot vaporization could only remove about half of the A molecules. Even though the stillpot is boiled up through the column repeatedly, the 'A' component never really gets to zero!
I owe you a huge sense of gratitude for stating that vacuum distillation must only be conducted using flawless glassware to prevent safety hazards. The owner of my local food factory plans to carry out the glycol reclamation process before Christmas. I hope he'll seek guidance from an expert to facilitate the entire thing. https://www.tegreclamationservices.com/